TIANSeq mRNA Capture Kit
Ominaisuudet:
■ Laaja valikoima näytteitä: Sopii mRNA: n sieppaamiseen korkealaatuisista (täydellisistä) näytteistä. RNA: n tulisi olla yli 7.
■ Kattavat tiedot: Täydelliset mRNA -tiedot säilytetään transkriptitietojen paikkansapitävyyden parantamiseksi.
■ Laaja käyttöalue: Soveltuu 100 ng-1 μg RNA: n sieppaamiseen.
Sovellukset
mRNA-kirjaston rakentaminen ja mRNA-sekvenssi.
Kaikki tuotteet voidaan räätälöidä ODM/OEM: lle. Yksityiskohtia varten,napsauta Mukautettu palvelu (ODM/OEM)




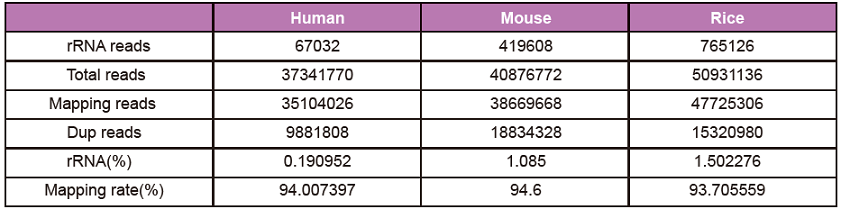
Taulukko 1. TIANSeq mRNA -kaappipaketin käyttäminen mRNA: n sieppaamiseen ihmisen, hiiren ja riisin kokonais -RNA: ssa syötetyn määrän ollessa 500 ng. RNA-Seq-kirjasto rakennettiin TIANSeq Fast RNA Library Kit (illumina) -laitteella, ja sekvensointitietojen analyysi osoitti, että kolmen tyyppisten näytteiden rRNA-tähde oli alle 2%ja sekvensointidatan yleinen laatu oli hyvä.
Tällä hetkellä suuren suorituskyvyn sekvensointitekniikka perustuu pääasiassa seuraavan sukupolven sekvensointitekniikkaan. Koska seuraavan sukupolven sekvensointitekniikan lukupituus on rajallinen, meidän on katkaistava täyspitkä sekvenssi pieniksi fragmenttikirjastoiksi sekvenssiksi. Eri sekvensointikokeiden tarpeiden mukaan valitsemme yleensä yksipäisen sekvensoinnin tai kaksipäisen sekvensoinnin. Tällä hetkellä seuraavan sukupolven sekvensointikirjaston DNA-fragmentit jakautuvat yleensä välillä 200-800 emäsparia.
a) DNA on huonolaatuinen ja sisältää inhibiittoreita. Käytä korkealaatuisia DNA-näytteitä välttääksesi entsyymiaktiivisuuden estämisen.
b) DNA-näytteen määrä ei ole riittävä, kun käytetään PCR-vapaata menetelmää DNA-kirjaston rakentamiseen. Kun sirpaleisen DNA: n syöttö ylittää 50 ng, PCR-vapaa työnkulku voidaan suorittaa valikoivasti kirjaston rakentamisprosessin aikana. Jos kirjaston kopiomäärä on liian pieni suoraan sekvensoimiseksi, DNA -kirjasto voidaan monistaa PCR: llä sovittimen ligaation jälkeen.
c) RNA -kontaminaatio johtaa virheelliseen DNA: n alkuperäiseen määritykseen RNA -kontaminaatio voi esiintyä genomisen DNA: n puhdistusprosessissa, mikä voi johtaa virheelliseen DNA: n kvantifiointiin ja riittämättömään DNA -lataukseen kirjaston rakentamisen aikana. RNA voidaan poistaa käsittelemällä RNaasilla.
A-1
a) Pienet palaset (60 bp-120 bp) ilmestyvät Pienet palaset ovat yleensä adapterifragmentteja tai sovittimien muodostamia dimeerejä. Puhdistus Agencourt AMPure XP -magneettisilla helmillä voi poistaa nämä sovitinpalat tehokkaasti ja varmistaa sekvensoinnin laadun.
b) Suuret fragmentit ilmestyvät kirjastoon PCR -monistamisen jälkeen Kirjasto -DNA -fragmentin koko kasvaa 120 bp: llä sovittimen ligatoinnin jälkeen. Jos DNA -fragmentti kasvaa yli 120 emäsparia adapterin ligaation jälkeen, se saattaa johtua epänormaalista fragmentin monistumisesta liiallisesta PCR -monistuksesta. PCR -syklien määrän vähentäminen voi estää tilanteen.
c) Kirjasto -DNA -fragmenttien epänormaali koko sovittimen ligaation jälkeen Tämän sarjan adapterin pituus on 60 bp. Kun fragmentin kaksi päätä liitetään sovittimiin, pituus kasvaa vain 120 bp. Jos käytät muuta kuin tämän sarjan mukana toimitettua sovitinta, ota yhteyttä toimittajaan ja anna tarvittavat tiedot, kuten sovittimen pituus. Varmista, että kokeilun työnkulku ja toiminta noudattavat oppaassa kuvattuja vaiheita.
d) Epänormaali DNA -fragmentin koko ennen sovittimen ligaatiota Tämän ongelman syy voi olla väärä reaktio -olosuhteet DNA: n pirstoutumisen aikana. Eri reaktioaikoja tulisi käyttää eri DNA -syöttöihin. Jos DNA-syöte on yli 10 ng, suosittelemme, että optimoinnin aloitusajaksi valitaan 12 minuutin reaktioaika, ja tällä hetkellä tuotettu fragmentin koko on pääasiassa välillä 300-500 bp. Käyttäjät voivat lisätä tai vähentää DNA-fragmenttien pituutta 2-4 minuutin ajan omien tarpeidensa mukaan optimoidakseen vaaditun koon DNA-fragmentit.
A-2
a) Hajoamisaikaa ei ole optimoitu Jos pirstoutunut DNA on liian pieni tai liian suuri, katso reaktioajan määrittämistä koskevista ohjeista fragmentaatioajan valintaohjeista ja käytä tätä ajankohtaa kontrollina ja aseta lisäksi reaktiojärjestelmä pidentää tai lyhentää 3 minuuttia, jotta voidaan säätää tarkemmin fragmentointiaikaa.
A-3
DNA: n epänormaali kokojakauma fragmentaatiokäsittelyn jälkeen
a) Fragmentointireagenssin väärä sulatusmenetelmä tai reagenssi ei ole täysin sekoittunut sulatuksen jälkeen. Sulata 5 × Fragmentation Enzyme Mix -reagenssi jäällä. Sulatuksen jälkeen sekoita reagenssi tasaisesti heiluttamalla varovasti putken pohjaa. Älä pyöritä reagenssia!
b) DNA -tulonäyte sisältää EDTA: ta tai muita epäpuhtauksia. Suola -ionien ja kelatointiaineiden ehtyminen DNA -puhdistusvaiheessa on erityisen tärkeää kokeen onnistumisen kannalta. Jos DNA liuotetaan 1 × TE: hen, suorita pirstoutuminen ohjeessa annetulla menetelmällä. Jos EDTA -pitoisuus liuoksessa on epävarma, on suositeltavaa puhdistaa DNA ja liuottaa se deionisoituun veteen myöhempää reaktiota varten.
c) Virheellinen DNA: n alustava kvantifiointi Hajanaisen DNA: n koko liittyy läheisesti syötetyn DNA: n määrään. Ennen fragmentaatiokäsittelyä DNA: n tarkka kvantifiointi Qubit-, Picogreen- ja muilla menetelmillä on välttämätöntä DNA: n tarkan määrän määrittämiseksi reaktiojärjestelmässä.
d) Reaktiojärjestelmän valmistelu ei noudata ohjeita. Hajanaisen reaktiojärjestelmän valmistelu on suoritettava jäillä tarkasti ohjeiden mukaisesti. Parhaan vaikutuksen varmistamiseksi kaikki reaktion komponentit on asetettava jäälle ja reaktiojärjestelmä on valmisteltava täydellisen jäähdytyksen jälkeen. Kun valmistus on valmis, pyyhkäise tai pipetoi sekoitaksesi huolellisesti. Älä pyöritä!
1. Väärä sekoitusmenetelmä (pyörre, väkivaltainen värähtely jne.) Aiheuttaa kirjastokappaleiden epänormaalin jakautumisen (kuten seuraavassa kuvassa), mikä vaikuttaa kirjaston laatuun. Siksi, kun valmistat Fragmentation Mix -reaktioliuosta, pipetoi varovasti ylös- ja alaspäin sekoittamiseksi tai käytä sormenpäätä heiluttamalla ja sekoittamalla tasaisesti. Varo sekoittamasta pyörteen kanssa.
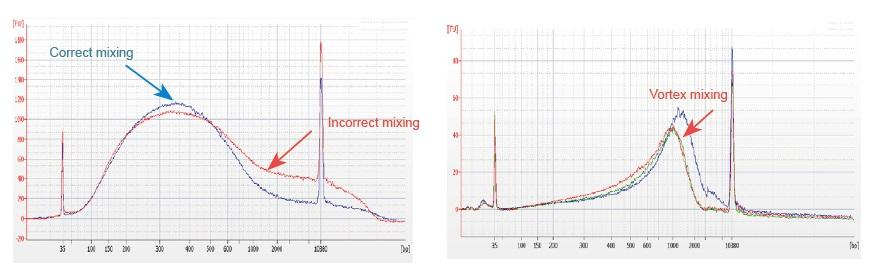
2. Kirjastojen rakentamiseen on käytettävä erittäin puhdasta DNA: ta
■ Hyvä DNA: n eheys: Elektroforeesikaista on yli 30 kb, ilman loppumista
■ OD260/230:> 1.5
■ OD260/280: 1,7-1,9
3. DNA -syötteen määrän on oltava tarkka. On suositeltavaa käyttää Qubit- ja PicoGreen -menetelmiä DNA: n kvantifioimiseksi Nanodropin sijasta.
4. EDTA -pitoisuus DNA -liuoksessa on määritettävä EDTA: lla on suuri vaikutus fragmentoitumisreaktioon. Jos EDTA -pitoisuus on korkea, DNA -puhdistus on suoritettava ennen seuraavaa testiä.
5. Fragmentointireaktioliuos on valmistettava jäällä. Fragmentointiprosessi on herkkä reaktiolämpötilalle ja -ajalle (erityisesti lisäaineen lisäämisen jälkeen). Reaktioajan tarkkuuden varmistamiseksi valmistele reaktiojärjestelmä jäällä.
6. Fragmentointireaktioajan on oltava tarkka Fragmentointivaiheen reaktioaika vaikuttaa suoraan fragmenttituotteiden kokoon ja vaikuttaa siten kirjaston DNA -fragmenttien kokojakaumaan.
1. Millainen näyte soveltuu tähän sarjaan?
Tämän pakkauksen soveltuva näytetyyppi voi olla kokonais -RNA tai puhdistettu mRNA, jolla on hyvä RNA -eheys. Jos kirjaston rakentamiseen käytetään kokonais -RNA: ta, on suositeltavaa käyttää rRNA: n poistopakettia (luettelo#4992363/4992364/4992391) rRNA: n poistamiseksi ensin.
2. Voiko FFPE -näytteitä käyttää kirjaston rakentamiseen tämän sarjan avulla?
FFPE -näytteiden mRNA hajoaa jossain määrin, suhteellisen heikko eheys. Kun käytät tätä sarjaa kirjaston rakentamiseen, on suositeltavaa optimoida pirstoutumisaika (lyhentää pirstoutumisaikaa tai olla suorittamatta pirstoutumista).
3. Mikä saattaa aiheuttaa lisätyn segmentin pienen poikkeaman tuotteen käyttöoppaassa annetun koonvalintavaiheen avulla?
Koko valitaan tarkasti tämän tuoteoppaan kokovalintavaiheen mukaisesti. Jos poikkeamia esiintyy, syy voi olla se, että magneettihelmet eivät ole tasapainossa huoneenlämpötilaan tai ne eivät ole täysin sekoittuneet, pipetti ei ole tarkka tai neste jää kärkeen. On suositeltavaa käyttää kokeessa vinkkejä, joiden adsorptio on vähäistä.
4. Sovittimien valinta kirjastorakentamisessa
Kirjastorakennussarja ei sisällä sovitinreagenssia, ja sitä on suositeltavaa käyttää yhdessä TIANSeq Single-Index Adapterin (Illumina) kanssa (4992641/4992642/4992378).
5. Kirjaston QC
Kirjaston kvantitatiivinen havaitseminen: Qubit ja qPCR käytetään määrittämään kirjaston massapitoisuus ja moolipitoisuus. Toiminta on ehdottomasti tuotteen käyttöohjeen mukaista. Kirjaston pitoisuus täyttää yleensä NGS -sekvensoinnin vaatimukset. Kirjaston jakelualueen havaitseminen: Agilent 2100 Bioanalyzerin avulla kirjaston jakelualueen havaitseminen.
6. Vahvistussyklin numeron valinta
Ohjeiden mukaan PCR-syklien lukumäärä on 6-12, ja tarvittavien PCR-syklien määrä on valittava näytetulon mukaan. Suurituottoisissa kirjastoissa ylivahvistusta esiintyy yleensä vaihtelevassa määrin, mikä ilmenee hieman suurempana huippuna tavoitealueen huipun jälkeen Agilent 2100 Bioanalyzerin havaitsemisessa tai havaittu Qubit-konsentraatio on pienempi kuin qPCR. Lievä ylivahvistus on normaali ilmiö, joka ei vaikuta kirjastojen sekvensointiin ja myöhempään data -analyysiin.
7. Piikit näkyvät Agilent 2100 Bioanalyzerin tunnistusprofiilissa
Piikkien esiintyminen Agilent 2100 Bioanalyzer -tunnistuksessa johtuu näytteiden epätasaisesta pirstoutumisesta, jolloin tietyn kokoisia fragmentteja tulee enemmän, ja tämä tulee selvemmäksi PCR -rikastamisen jälkeen. Tässä tapauksessa on suositeltavaa olla tekemättä kokovalintaa, eli asettaa fragmentaatio -olosuhteeksi 94 ° C 15 minuutin inkuboinnin ajaksi, jolloin fragmentin jakauma on pieni ja väkevä ja homogeenisuutta voidaan parantaa.
Tuoteryhmät
MIKSI VALITA MEIDÄT
Tehtaamme on perustamisestaan lähtien kehittänyt maailmanluokan tuotteita periaatteen mukaisesti
ensin laatu. Tuotteemme ovat saaneet erinomaisen maineen alalla ja arvostusluottamuksen uusien ja vanhojen asiakkaiden keskuudessa.
- Puh: +86010-59822688
- Rakennus 5, nro 86, Shuangying West Road, Changping District, Peking.
- people@tiangen.com



